

Crouzon syndrome is a autosomal dominant genetic disorder called a branchial arch syndrome. Specifically, this syndrome affects the first branchial (or pharyngeal) arch, which is the precursor of the maxilla and mandible. Considering that the branchial arches are significant developmental characteristics in a expanding embryo, disturbances within their own evolution create lasting and widespread results.
This syndrome is called after Octave Crouzon, a French doctor who first described this ailment. He noticed that the affected patients were also a mom and her daughter, indicating a genetic foundation. First called “craniofacial dysostosis”, the disease was characterized by numerous clinical features. This syndrome results from a mutation in the fibroblast growth factor receptor II, located on chromosome 10.
Breaking down the name, “craniofacial” identifies the skull and face, and “dysostosis” describes malformation of bone. Currently called Crouzon syndrome, the traits can be clarified by the basic significance of its former title. What happens is that a baby’s skull and facial bones, whereas in evolution, fuse premature or cannot expand. Therefore, normal bone development cannot happen. Fusion of different sutures results in distinct patterns of expansion of the skull.
Signs and symptoms of crouzon syndrome
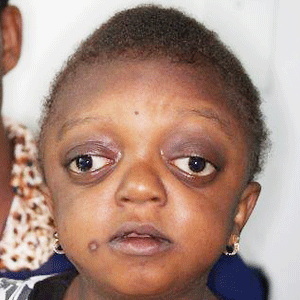
As a result of the alterations to the developing embryo, the indicators are extremely conspicuous features, particularly in the face. Low-set ears are a standard feature, as in each one of the ailments that are known as branchial arch syndromes. The cause of the abnormality is that ears onto a foetus are considerably lower compared to those on a grownup. During normal growth, the ears “traveling” up on the mindnonetheless, at Crouzon patients, that this pattern of growth is interrupted. Ear canal malformations are incredibly common, generally leading to some hearing loss. In especially severe cases, Ménière’s disorder might happen.
The most noteable feature of Crouzon syndrome is craniosynostosis, as explained previously; nevertheless it generally presents as brachycephaly leading to the look of a brief and broad mind. Furthermore, outside strabismus is a frequent occurrence, which may be considered as opposite in the eye position located in Down syndrome. Last, hypoplastic maxilla (inadequate rise of the midface) contributes to comparative mandibular prognathism (chin seems to float despite an ordinary rise of mandible) and provides the consequence of the individual using a concave face.
The most notable feature of Crouzon syndrome is craniosynostosis, as explained previously; nevertheless, it generally presents as brachycephaly leading to the look of a brief and broad mind. Furthermore, outside strabismus is a frequent occurrence, which may be considered as opposite in the eye position located in Down syndrome. Last, hypoplastic maxilla (inadequate rise of the midface) contributes to comparative mandibular prognathism (chin seems to float despite an ordinary rise of mandible) and provides the consequence of the individual using a concave face.
Causes of crouzon syndrome
The current research indicates fibroblast growth factor receptors (FGFR) FGFR2 and FGFR3 as the leading factors in causing the autosomal dominant Crouzon Syndrome] These two transmembrane proteins are two of four fibroblast growth factor receptors involved in osteoblast differentiation during embryonic development; mutations amongst these receptors are involved in several genetic disorders. There are 40 known mutations, the majority of which are brought on by a missense mutation.FGFR2 is the most frequently mutated gene, a missense in cysteine 342 in exon 9, which produces a gain-of-function. The FGFR2lllc isoform, generated via alternative splicing of exon 3 of the FGFR2 gene, utilizes exon 9 and can be employed in mesenchymal stem cells to restrain ossification. On the other hand, the mutation constitutively activates the transmembrane protein using a disulfide bond formed wrongly as a result of a reduction of cysteine 342. FGFR3 is expressed more from the frontal bones through embryonic development, directing cranial bone growth. A point mutation induces constitutive activation of tyrosine from the activation loop, found in the cytosolic region of the protein, resulting in the accelerated distinction of rectal osteoblasts. Resulting in an early fusion of rectal-cranial bones.

